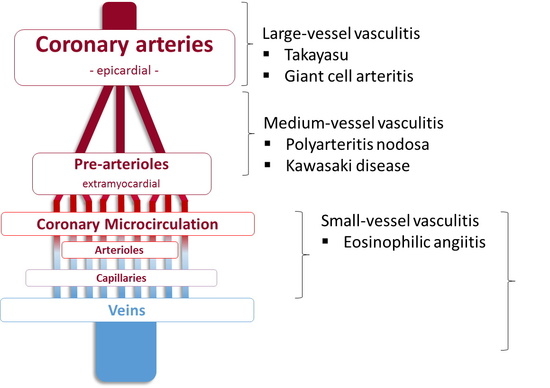
El síndrome de Behçet es una enfermedad autoinmune sistémica que se incluye dentro de las vasculitis, afecciones en las que se produce una inflamación de los vasos sanguíneos de causa desconocida, que puede afectar a casi cualquier parte del organismo. Se trata de una enfermedad poco frecuente, que suele debutar entre los 20 y 40 años, y su prevalencia en España se cifra entre 5 y 10 casos por 100.000 habitantes.
“Esta patología provoca unas lesiones características en la piel y las mucosas. Además, con frecuencia, ocasiona alteraciones en los ojos (uveítis) y en las articulaciones; y más rara vez causa inflamación intestinal, trombosis de venas, dilataciones arteriales e inflamación del sistema nervioso”, según explica el Dr. Jenaro Graña, reumatólogo del Complejo Hospitalario Universitario de A Coruña.